Enols and Enolates
Keto-Enol Tautomerism
Last updated: February 12th, 2025 |
Keto-Enol Tautomerism
Many ketones and aldehydes have an alter-ego “enol” form with completely different chemical properties than the familiar “keto” form. In this article we’ll explore the structure and properties of this “enol” form, go through the mechanism for the keto-enol transformation, and describe some of the key factors that can affect the keto-enol equilibrium.
*mostly aldehydes and ketones, although it can occur in other species as well (e.g. acid halides)
NOTE: This is a 2022 update of an older post entitled, “Keto-Enol Tautomerism: Key Points”.
Table of Contents
- When Ketones Moonlight As Nucleophiles
- Keto-Enol Tautomerism
- Examples Of Keto-Enol Tautomerism
- Properties of Enols
- Keto-Enol Tautomerism: Mechanisms
- Four Factors That Affect Keto-Enol Equilibria
- Factor #1: Substitution
- Factor #2: Conjugation/Resonance
- Factor #3: Hydrogen Bonding
- Factor #4: Aromaticity
- Revisiting A Weird, “Unketone-like” Reaction
- Summary And Conclusion
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. When Ketones Moonlight As Nucleophiles
Reactions of aldehydes and ketones: by this point we’ve pretty much seen ’em all. Right?
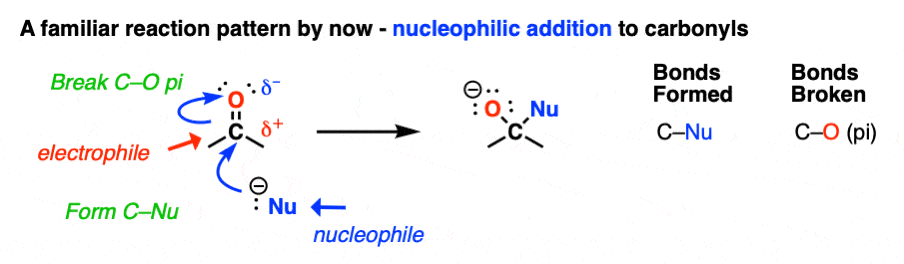
I mean, the carbonyl carbon of aldehydes and ketones is an electrophile. So their reactions all follow the same pattern, more or less.
- A nucleophile attacks carbon, forming C–Nu and breaking the C-O pi bond to give an alkoxide (O–) This is nucleophilic addition.
- Mild acid is added to protonate the alkoxide and form O–H. [See here: Aldehydes and Ketones – 14 Reactions With The Same Mechanism ]
So that’s all there is to the reactions of aldehydes and ketones. Case closed?
Not quite. This is the article where we discover that most aldehydes and ketones live a double life.

By day, they are respectable electrophiles, undergoing addition with nucleophiles. But by night, maybe when the moon is full, they undergo a transformation into a completely different beast – one with a different structure, properties… and appetites.
Here’s an example of a reaction which doesn’t fit the normal “two-step” pattern. Treating a ketone with acid and Br2 gives…. a new C-Br bond next to the carbonyl carbon?
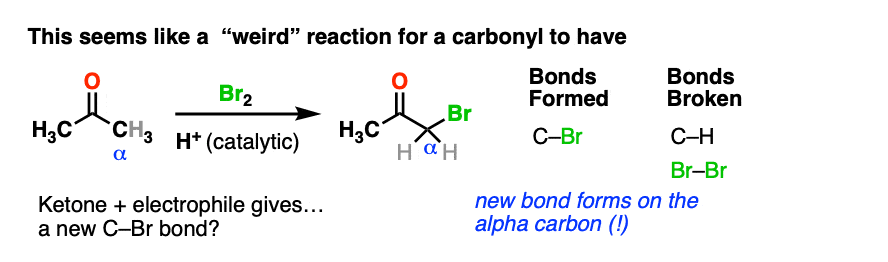
This seems decidedly un-ketone-like. Recall that Br2 is electrophilic (remember how it reacts with electron-rich alkenes and aromatic rings).
So what the heck is our “electrophilic” ketone doing cavorting around with another electrophile? And forming bonds at…. a totally different place?
If Br2 is the electrophile, then what was the nucleophile here?
2. Keto-Enol Tautomerism
A clue to this aberrant chemical behavior is provided not by lycanthropy, but by the fact that many aldehydes and ketones are in equilibrium with a structural isomer known as the enol form (part alkene, part alcohol).
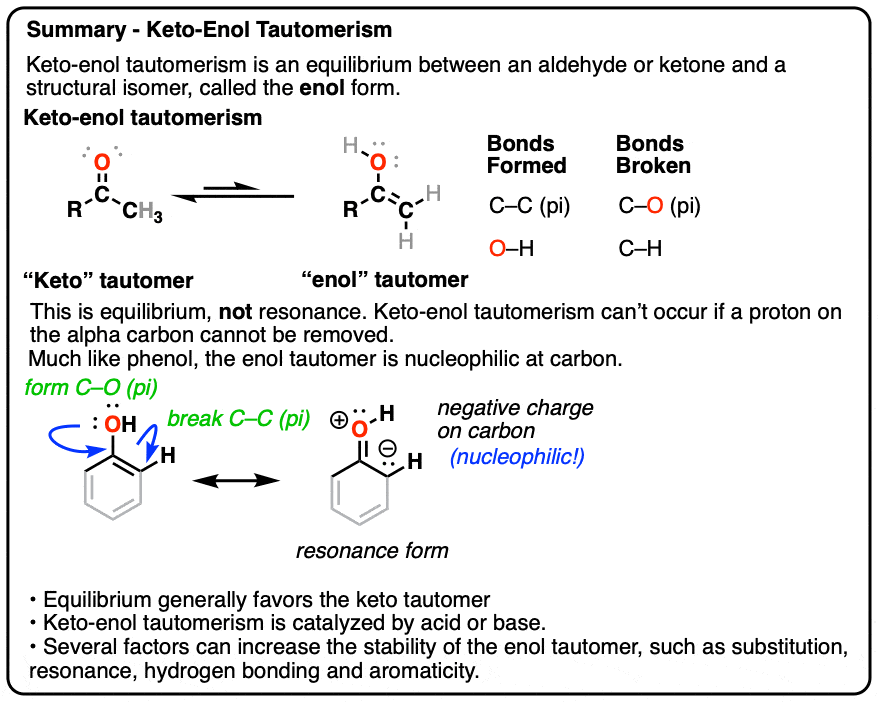
This behavior is known as keto-enol tautomerism.
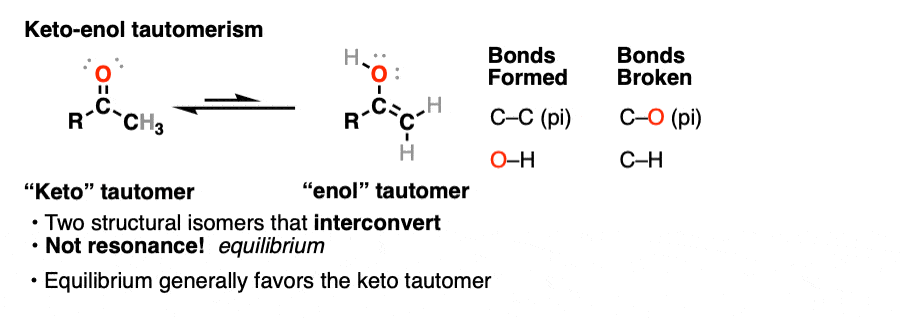
The keto and enol forms are not resonance forms! [Note 1 ] They are structural isomers that can interconvert. This property is called tautomerism. Keto-enol tautomerism is the most commonly-encountered type of tautomerism, although there are others. [Note 2]
That means that the keto and enol forms, if separated, have very different physical and chemical properties, which will become important in a moment. [Note 3]
We’ve actually encountered keto-enol tautomerism before, if only briefly. You may recall that alkynes treated with HgSO4 and water give an enol intermediate, which then tautomerizes to a ketone. Similarly, alkynes undergo hydroboration-oxidation to give enols which can tautomerize to aldehydes. [See Hydroboration and Oxymercuration of Alkynes]
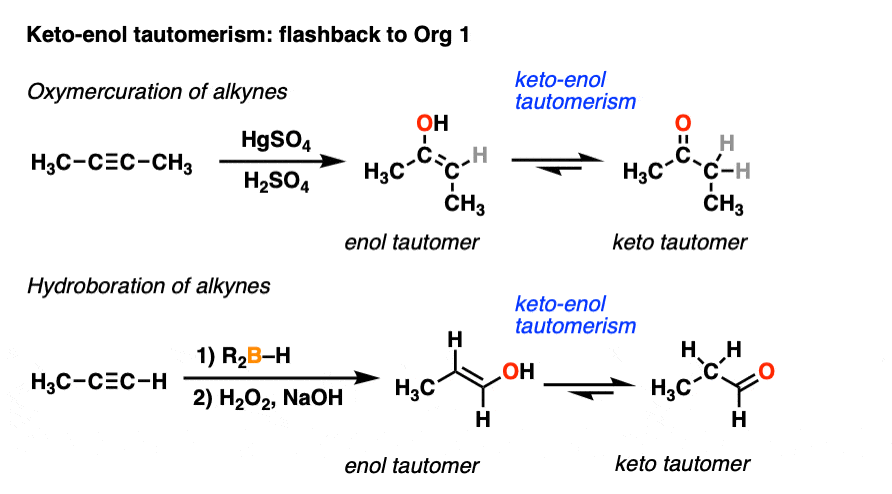
Your instructor may have glossed over the tautomerism part at the time, saying something like, “you’ll learn more about this in Org 2.” Well, the moment has arrived.
3. Some Examples of Keto-Enol Tautomerism
Here are a few more examples of keto-enol tautomerism.
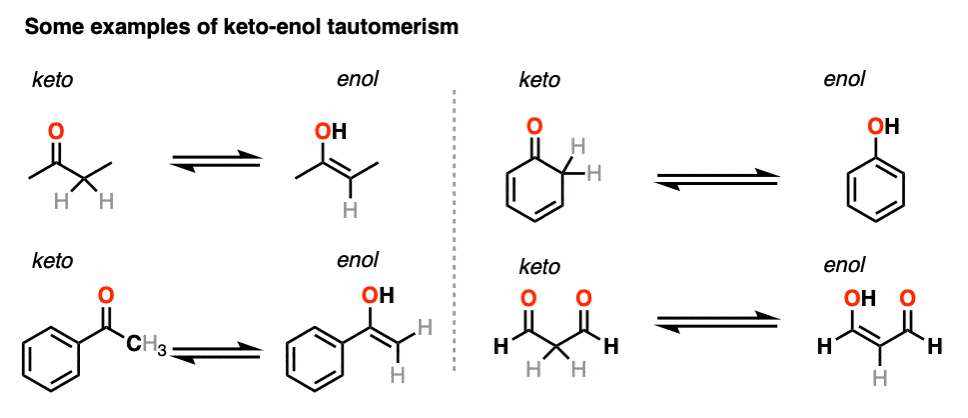
You might recall that at the very beginning I said that keto-enol tautomerism happens in some aldehydes and ketones. Why “some” but not all? Because it can’t happen in cases where there is no hydrogen on the alpha carbon. [Note 4]
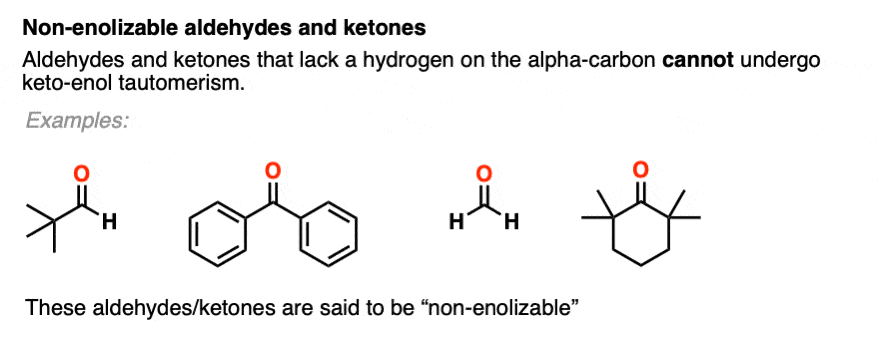
These aldehydes and ketones are known as “non-enolizable” aldehydes and ketones.
4. Properties of Enols
Might this enol form be responsible for those “weird” reactions of aldehydes and ketones we were talking about?
[NARRATOR: You think??? Why else would he be bringing this up?]
A closer look at the enol form may help us to understand some of its properties.
In the enol form, a lone pair on oxygen is in conjugation with the C-C pi bond. [See post: Conjugation and resonance]
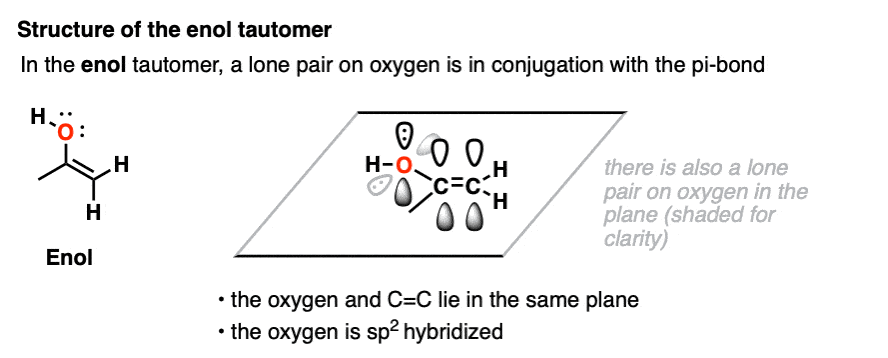
At first glance, we might think that having an electronegative oxygen attached to an alkene might make it more electron-poor, since it’s sucking electron density away through that C–O sigma bond.
However, we can also draw a resonance form where a lone pair on oxygen attached to the pi-bond can donate a pair of electrons towards the ring, forming a new C–O pi bond.
In the process, the pair of electrons in the C–C pi bond moves over to become a lone pair on the adjacent carbon.
This is known as pi-donation and has the effect of making an attached pi bond more electron-rich and therefore more nucleophilic. [See post: Pi-Donation]
On balance, the pi-donation effect from oxygen tends to be much stronger than the inductive effect arising from electronegativity.
You may recall that OH is a strongly activating group for electrophilic aromatic substitution. [See: Activating and Deactivating Groups] You might even recall drawing resonance forms which show the electron density moving towards the ortho- and para- positions.
If you remember that, I’ve got good news. Enols behave in exactly the same way!
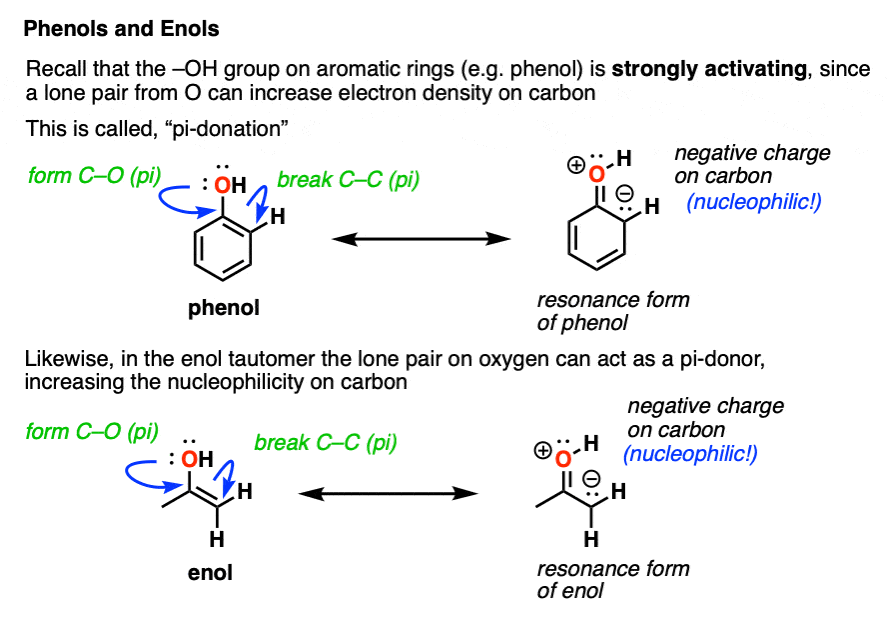
An attached -OH group has the effect of making the carbon adjacent to the C–O bond more electron rich and therefore more nucleophilic!
This helps to untangle the mystery of why the new C–Br bond was formed on the “alpha carbon” in our example above. In an enol, the alpha-carbon is strongly nucleophilic!
5. Keto-Enol Tautomerism: Mechanisms
This begs a question. If the keto and enol tautomers aren’t resonance forms, then there must be some process by which they interconvert. So how does this happen?
It might be instructive to show how to do it the wrong way first.
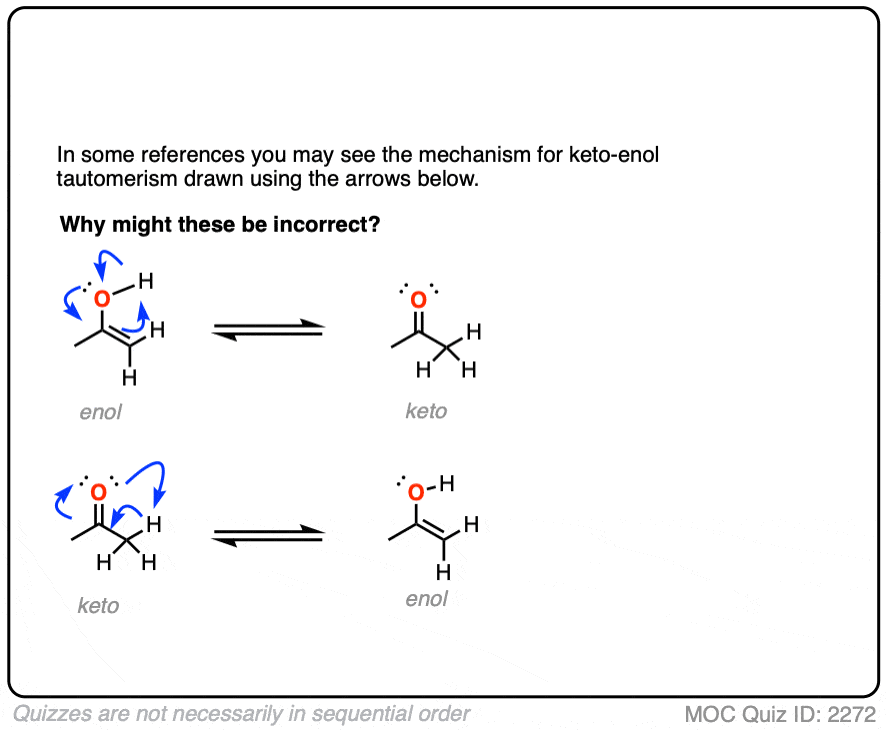 Click to Flip
Click to Flip
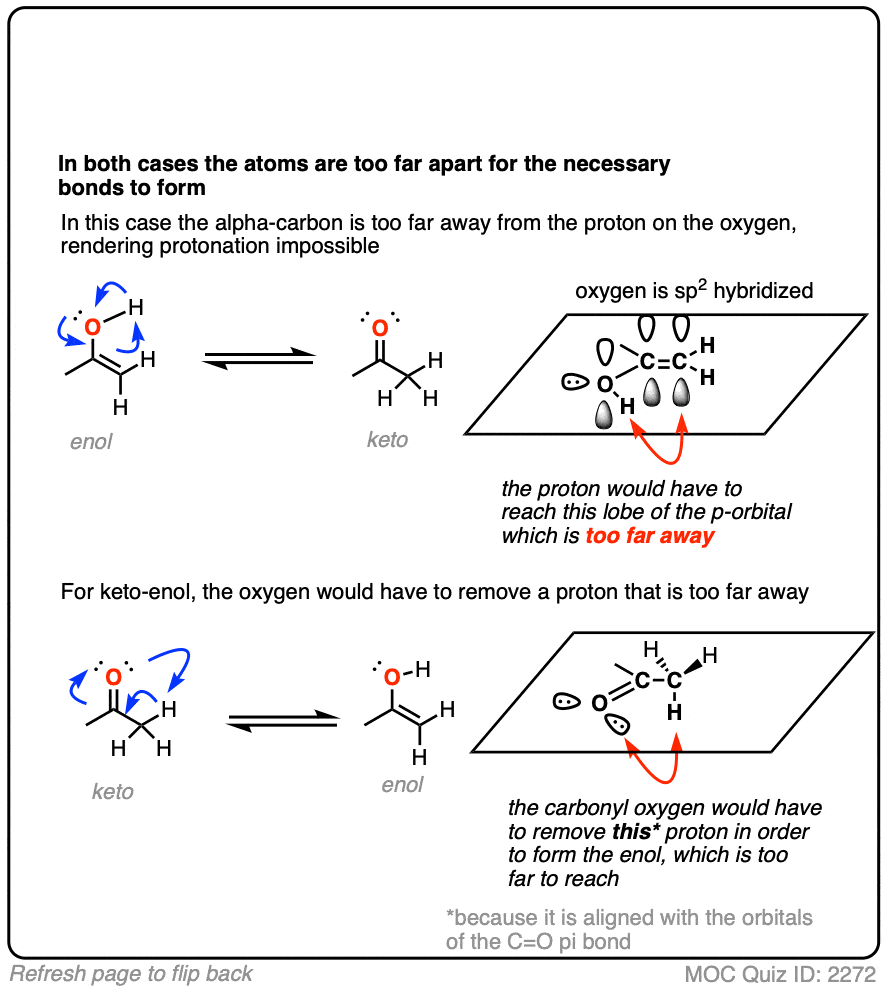
Hold on. It looks so gosh-darn tempting to draw it all up as one concerted process. How can this possibly be wrong?
It’s wrong for the same reason that you can’t scratch your left elbow with your left hand. They are just too far apart to touch.
What’s needed is a “helper” molecule to transport the proton from one part of the molecule to the other. Water serves nicely. [Note 5]
In addition to water, interconversion of keto and enol tautomers is greatly assisted by the presence of acid or base. [Note 6]
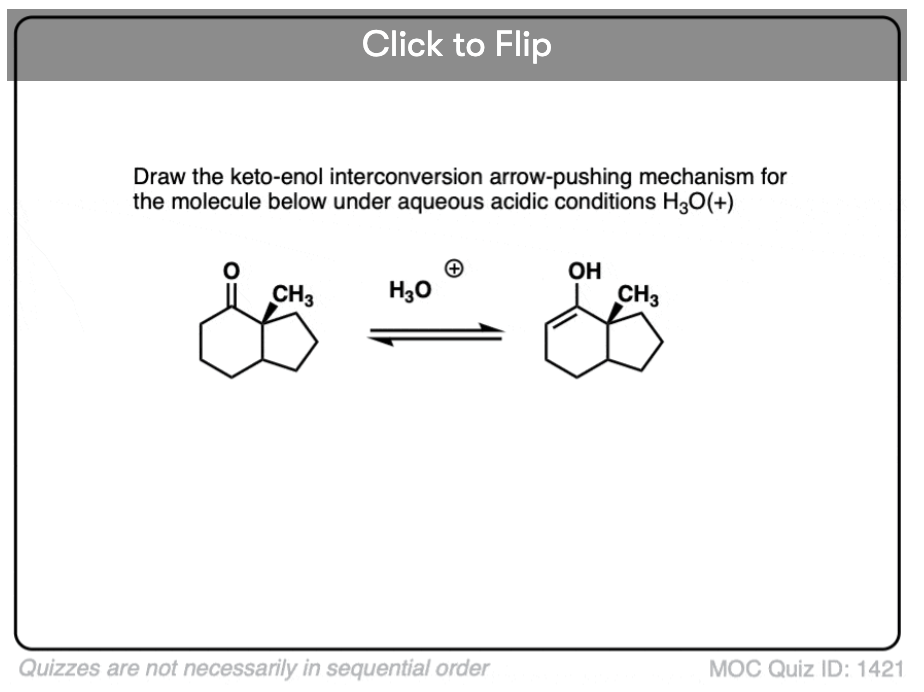
In the presence of acid, the carbonyl oxygen is protonated (Step 1, form O-H). Then, in the slow step, the alpha-carbon is deprotonated to give the enol (Step 2, break C-H, form C-C (pi), break C-O (pi). ). This gives us the enol.
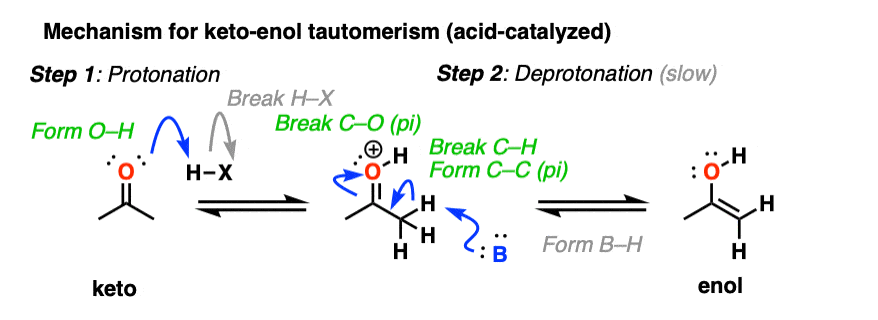
For the acid-catalyzed conversion of enol to keto, hover here and an image will pop up or click on this link.
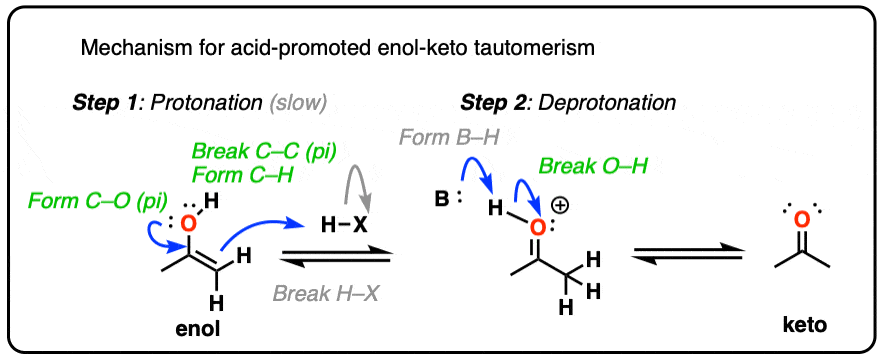{kind=link}
Keto-enol interconversion is also assisted by base. Here, deprotonation of the alpha carbon (Step 1, break C-H, form C-C (pi), break C-O pi) is the slow step, and protonation of the oxygen (Step 2, form O-H) is the fast step.
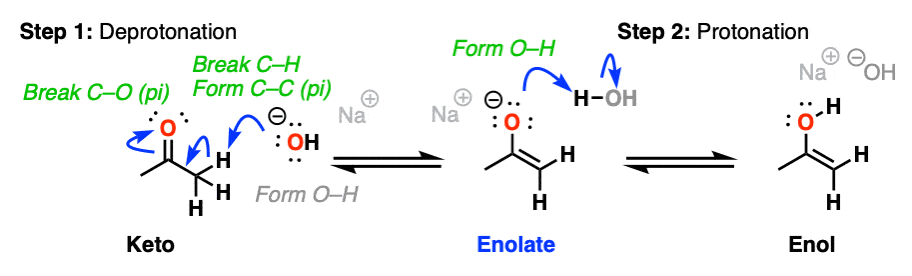
(The conjugate base of an enol is called an enolate. We’ll have a lot more to say about enolates shortly).
For the reverse process (base-catalyzed conversion of enol to keto) hover here or click on the link.
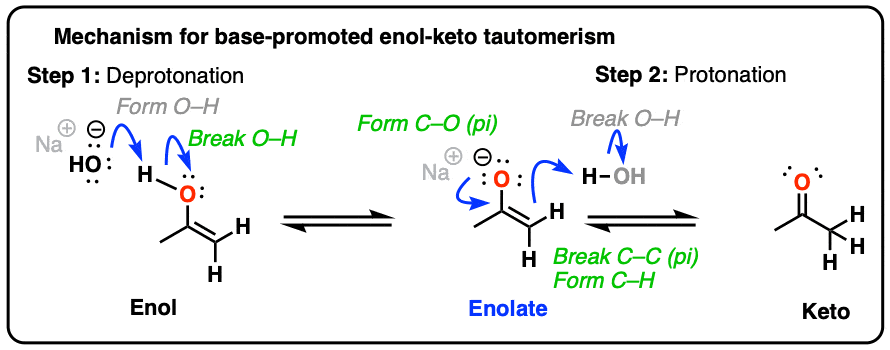{kind=link}
6. Four Factors That Affect Keto-Enol Equilibria
For most aldehydes and ketones, equilibrium strongly favors the keto form, often by a factor of 104 or more. This mostly has to do with the difference in bond strengths (C-O pi is a stronger bond than C–C pi , for more details see Note 7 ). The enol form is even less favored for carboxylic acids and esters. [Note 8]
That said, there are at least 4 key factors that can significantly influence the keto: enol ratio. It’s important to know them as they make good exam question material.
They are, in order from weakest to strongest influence:
- substitution – enols are a type of alkene, and substituted alkenes are more stable
- conjugation – conjugation of the enol pi-bond with a neighboring pi system is stabilizing
- hydrogen bonding – intramolecular hydrogen bonding can stabilize the enol form
- aromaticity – if the enol is part of an aromatic ring, expect the enol form to dominate
Factor #1: Substitution
Remember Zaitsev’s rule? Eliminations tend to favor formation of the more substituted alkene, because they are more thermodynamically stable? [See: Stability of Alkenes]
The same trend applies to enols!
For example, which enol do you think will be favored at equilibrium?
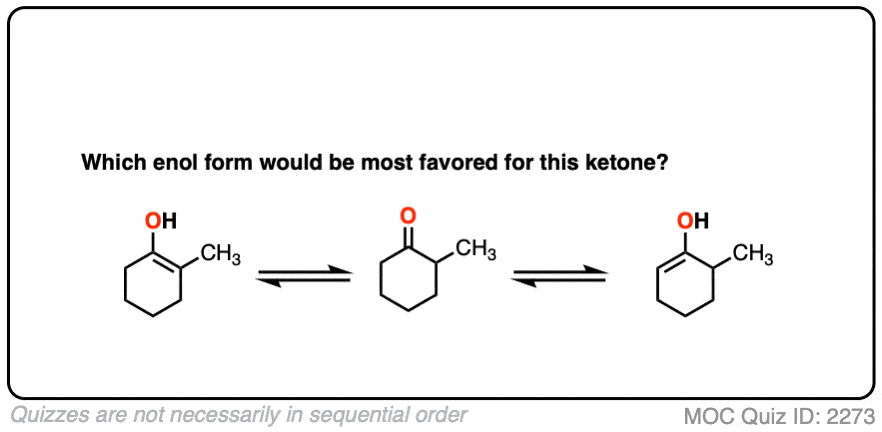 Click to Flip
Click to Flip
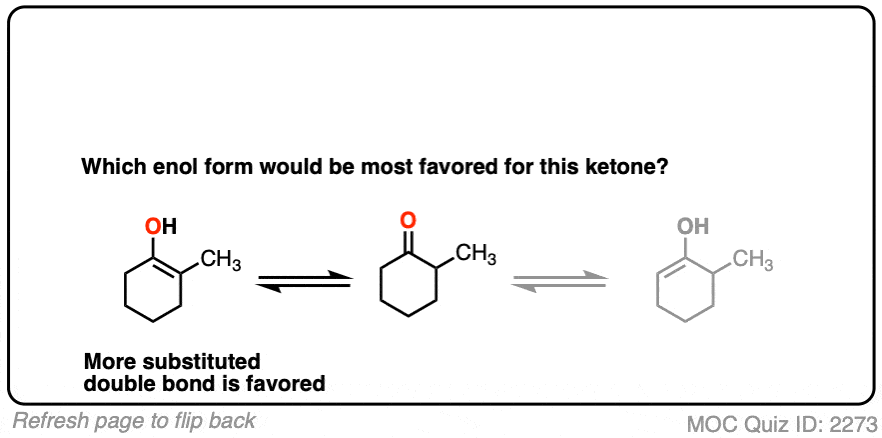
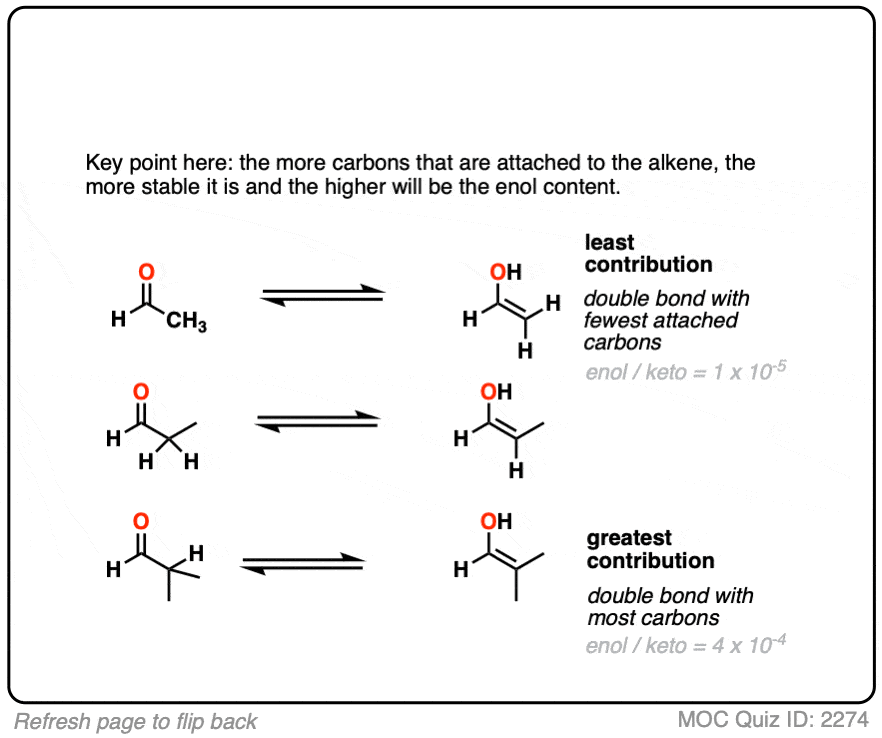
The more substituted an enol is, the more thermodynamically stable it tends to be. The difference often isn’t huge (1-2 kcal/mol) but recall that even a difference of 1 kcal/mol means an equilibrium ratio of about 80:20. [Calculated here]
(This is actually quite an important trend to be aware of, because you may soon be contrasting kinetic versus thermodynamic enolates.)
Compare the stability of the following aldehyde enols:
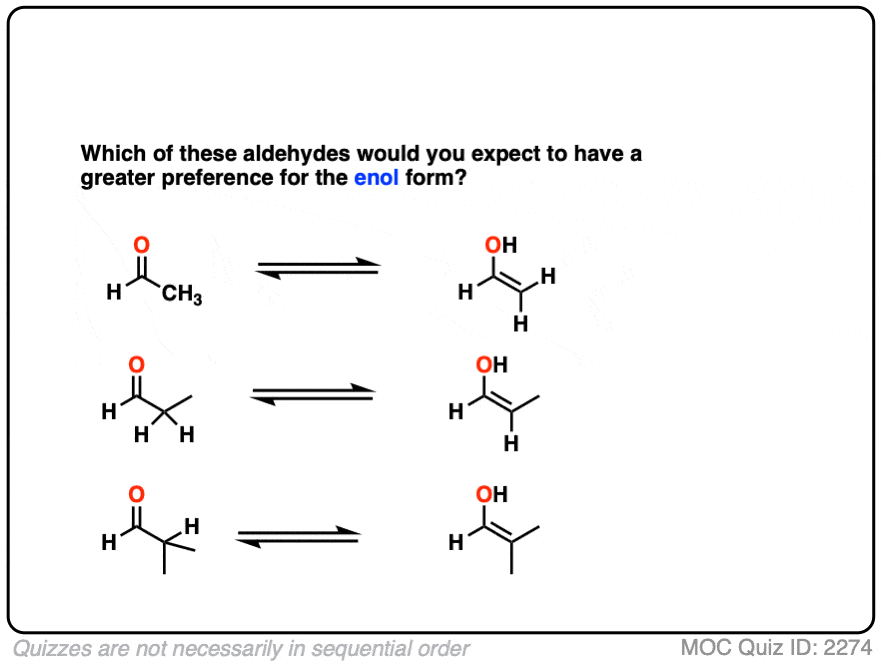 Click to Flip
Click to Flip
Factor #2: Conjugation / Resonance
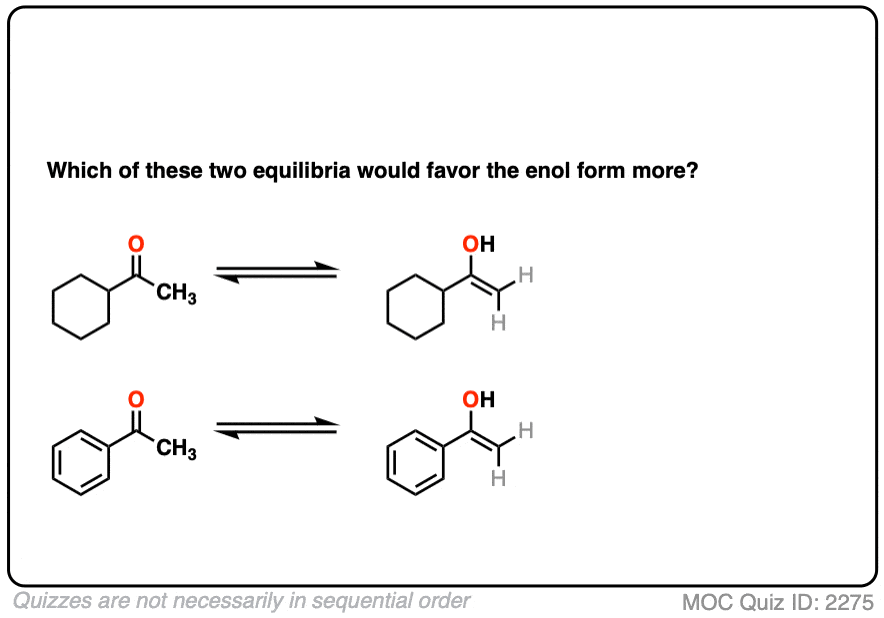 Click to Flip
Click to Flip
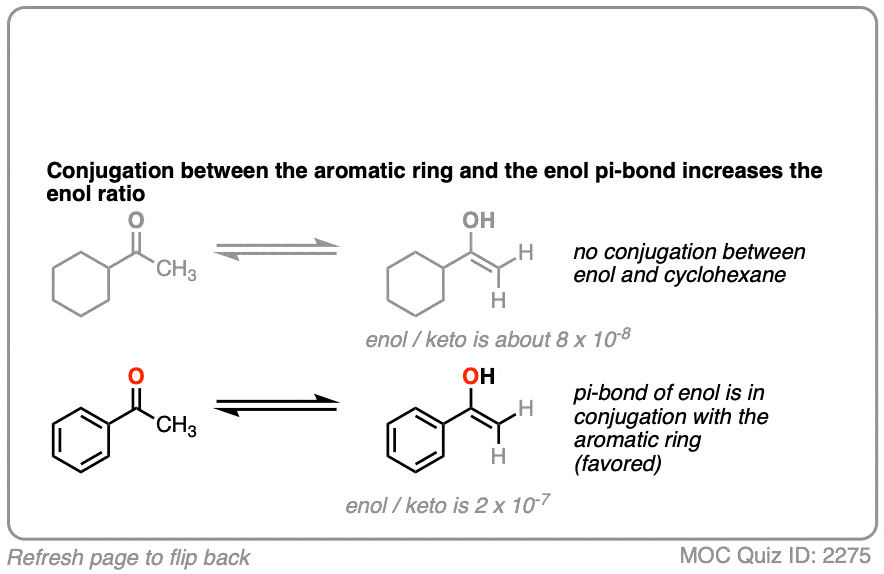
π bonds are a little like Cheerios in milk: it’s more stable for them to connect together rather than hang out in isolation.
Which leads us to ask: which of these two ketones will have a greater preference for the enol form?
 Click to Flip
Click to Flip
The more favorable enol form will be that which allows conjugation with a neighboring pi system.
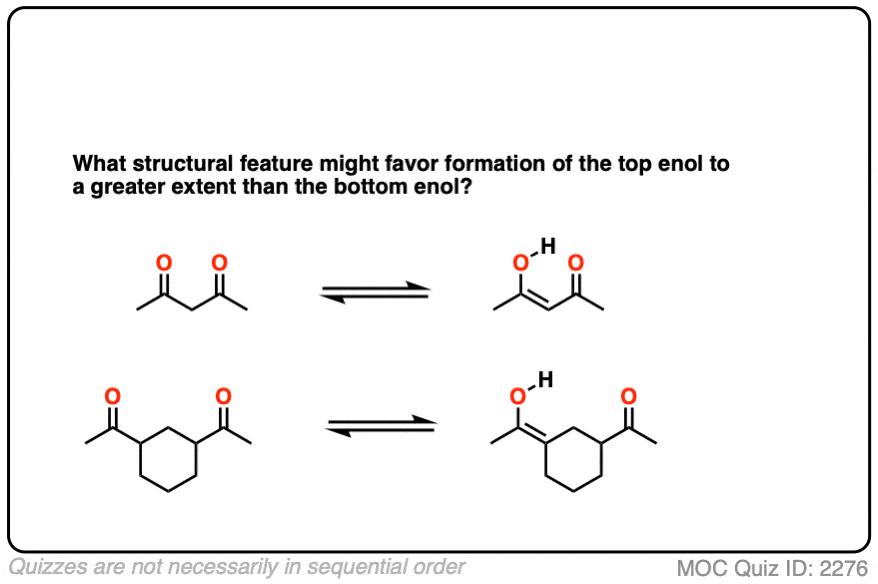
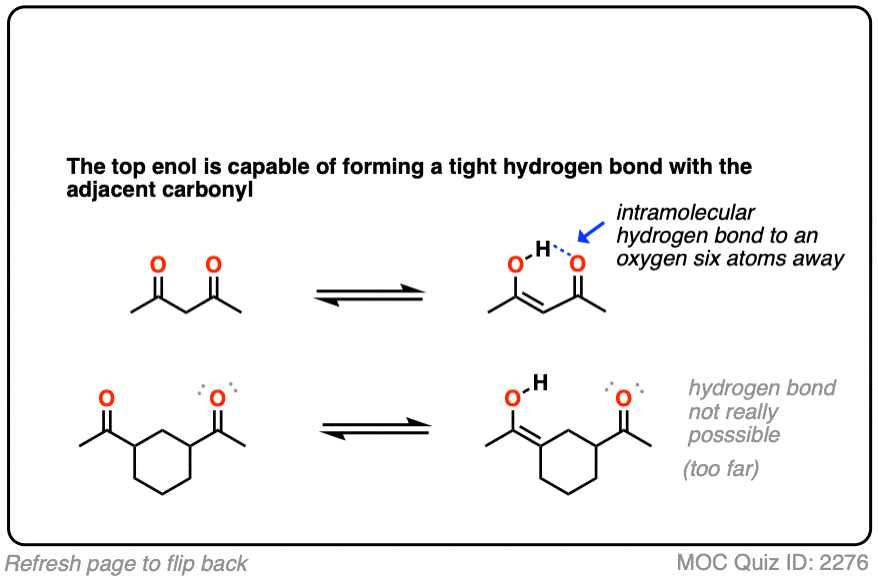
Factor #3: Hydrogen Bonding
This is an interesting one. Enols have an O-H bond, which is highly polarized; the hydrogen bears a partially positive charge and is capable of hydrogen bonding. If a Lewis base (e.g. the oxygen of a carbonyl) is present nearby, an intramolecular hydrogen bond can result, which will stabilize the enol form.
 Click to Flip
Click to Flip
In some cases the proportion of enol form can be equal to or even greater than the amount of keto tautomer in solution.
(An NMR spectrum of 2,4-pentanedione, for example shows about a 1:1 mixture of the keto and enol tautomers) hover for image
[There is some dependence on solvent here. See Note 9]
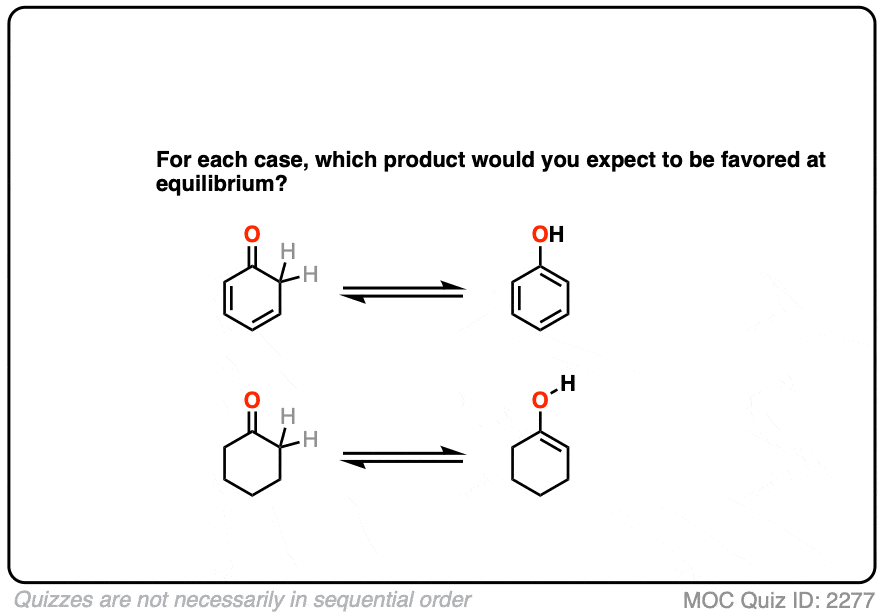
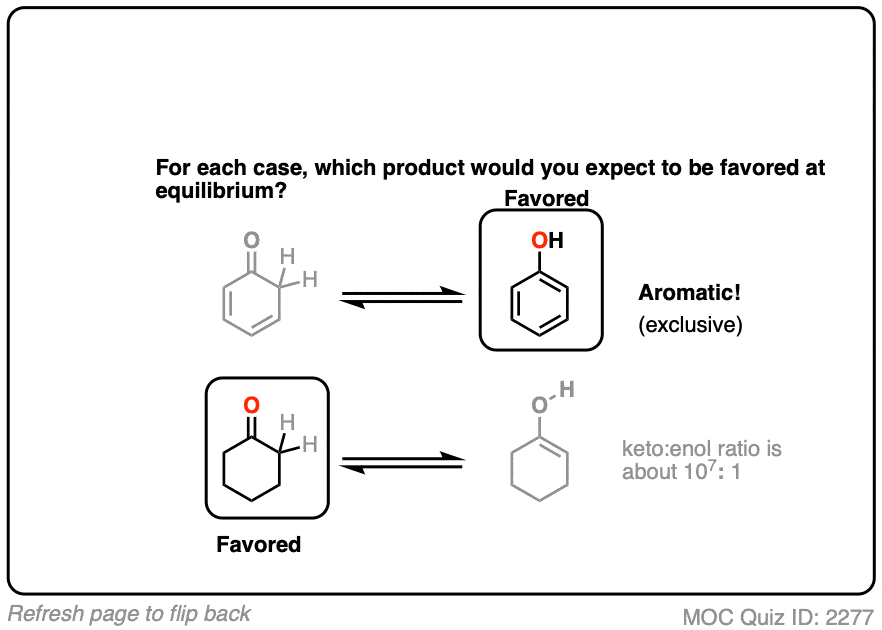
Factor #4 : Aromaticity
The most powerful driving force to favor an enol tautomer is aromaticity.
Contrast these two ketones. Which would favor the enol tautomer more?
 Click to Flip
Click to Flip
Hopefully you can see that the aromaticity of phenol (with its resonance energy of >20 kcal/mol) greatly favors the enol form here. The keto tautomer can’t be detected in solution!
11. Revisiting An “Un-Ketone-Like” Reaction With Br2
After all we’ve explored, I think we’re ready to go back at this “weird” reaction of a ketone one more time.
How might this reaction work?
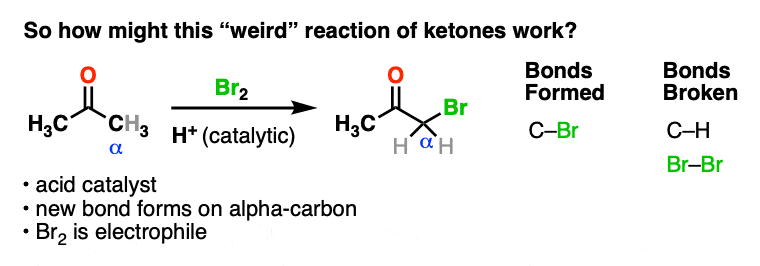
We’re adding acid and forming a new bond to an electrophile at the alpha carbon. I think we can safely say that the first step is acid-catalyzed keto-enol tautomerism.
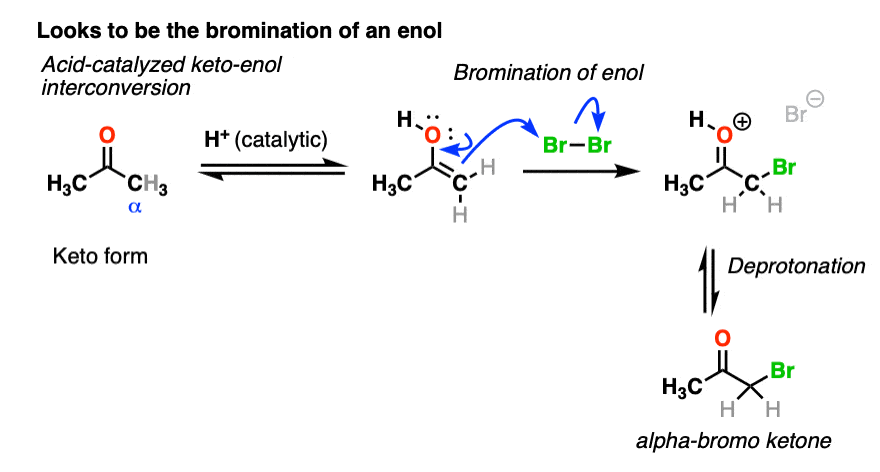
This gives us a nucleophilic enol, which can then react with Br2 to form a new C-Br bond. Deprotonation then gives the neutral alpha-bromo ketone.
12. Summary and Conclusion
So what have we learned?
- Aldehydes and ketones that have a proton on the alpha carbon can participate in keto-enol tautomerism, where an equilibrium exists between two constitutional isomers – the keto and enol forms. Constitutional isomers that are in equilibrium are called “tautomers”.
- The enol tautomer is nucleophilic on the carbon adjacent to the C-OH bond (the “alpha-carbon”) and undergoes reactions with electrophiles
- Keto-enol tautomerism can be catalyzed with acid or base.
- Generally the keto tautomer is favored at equilibrium.
- Many structural factors that stabilize alkenes such as substitution, conjugation, and participation in an aromatic ring may also help to stabilize the enol form. Also, the enol tautomer can be stabilized by a neighboring hydrogen bond acceptor (such as a ketone)
Notes
Some representative keto-enol ratios (collected from Carey & Sundberg and March’s Advanced Organic Chemistry)
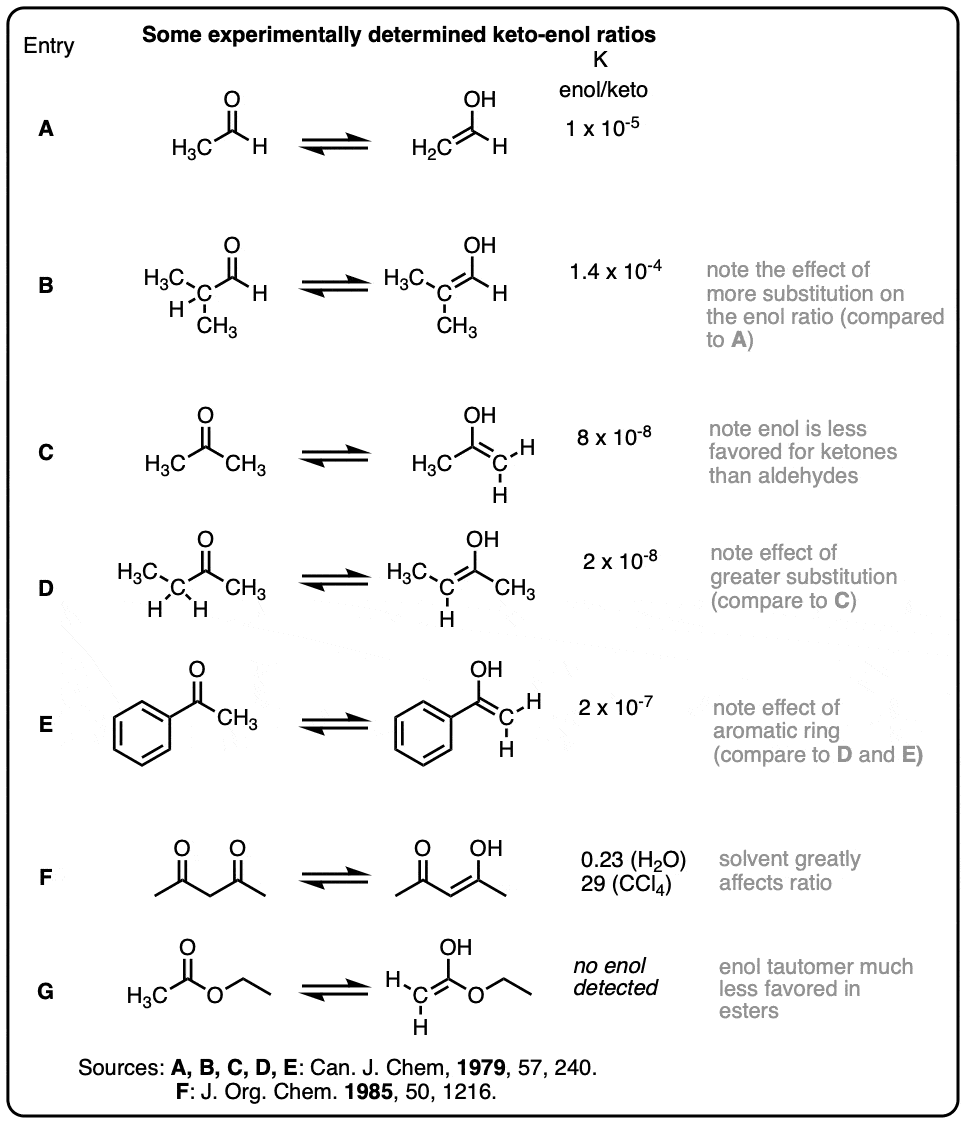
Note 1 – Recall that resonance forms are not in equilibrium with each other – they are just ways of representing the distribution of pi-electrons in a molecule whose true structure is best thought of as a weighted hybrid of resonance forms. [See: Resonance Structures]
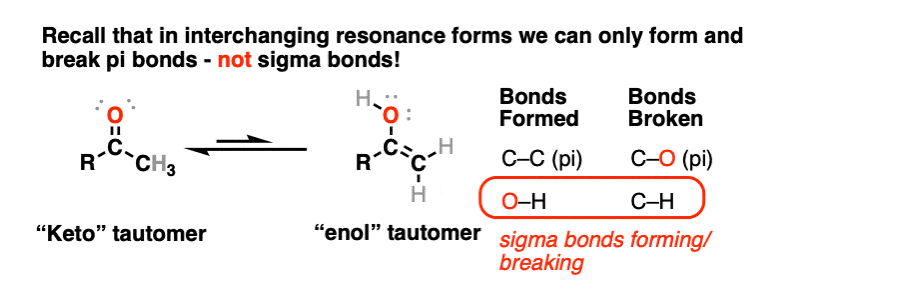
In the transformation of a keto- to an enol- form, a C-H sigma bond and a C-O pi bond breaks, and an O-H sigma bond and C-C pi bond forms. This is another reason why these can’t be resonance forms – recall that we can’t break sigma bonds to interconvert resonance structures.
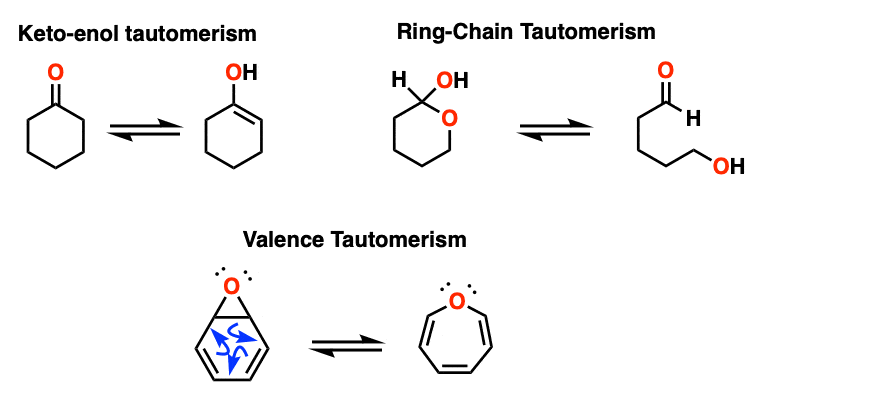
Note 2 – Keto-enol tautomerism is one of the most prominent types of tautomerism. Ring-chain tautomerism, which often happens in sugars, is another. [See: Ring Chain Tautomerism In Sugars]. A third is valence tautomerism, which occurs in certain molecules that lack a fixed structure.
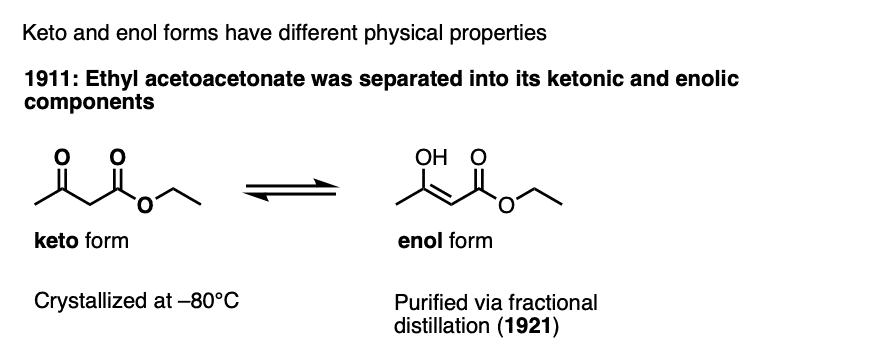
Note 3 – The keto and enol forms have been separated for certain ketones. The first example was ethyl acetoacetate.
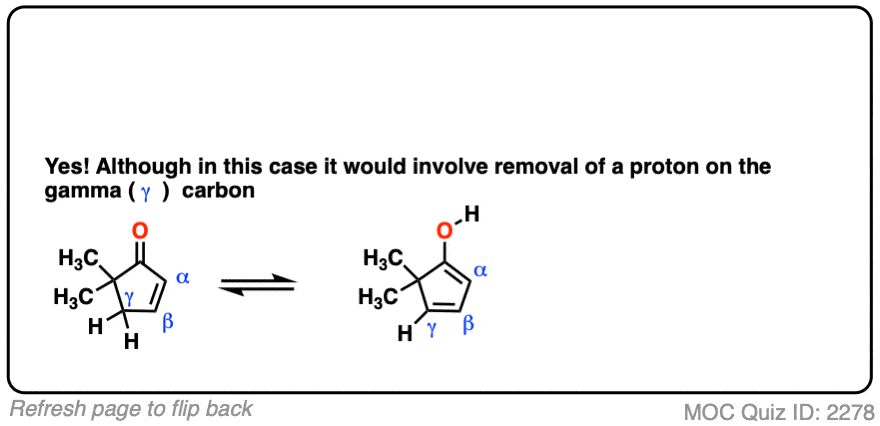
Note 4 – It is possible for certain alpha, beta unsaturated ketones that appear “non-enolizable” to be enolized at the gamma position.
Click to Flip
Note 5- It is possible in certain cases to prepare “pure” enols, uncontaminated with their keto tautomer. In these cases researchers are extremely careful to exclude water, which may act as a “proton shuttle” to interconvert the enol and keto forms.
Note 6 – See Ref 1 for more details on how acid accelerates the rate of keto-enol tautomerism.
Note 7 – A quick tabulation of the bonds that form and break using average values for their bond dissociation energies reveals at least a 10 kcal/mol difference between the keto and enol tautomers in most cases favoring the keto. See this quiz for an example.
Note 8. The proportion of enol tautomer in a solution of ethyl acetate was found to be less than one part in 10 million – several orders of magnitude below that found for ketones.
Note 9. There is some solvent dependence on the keto:enol ratio. In a non hydrogen-bonding solvent (like carbon tetrachloride) the enol:keto ratio for ethyl acetoacetate was measured to be around 98:2 . In a hydrogen-bonding solvent like water, however, there is more “competition” for accepting a hydrogen bond, and the enol: keto ratio drops to about 1 : 4. (See Ref 5)
Quiz Yourself!
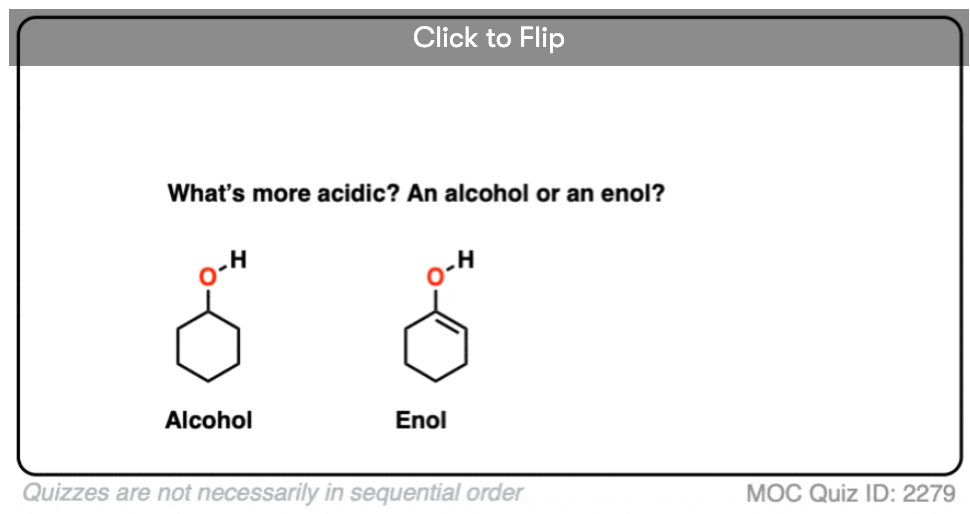
Become a MOC member to see the clickable quiz with answers on the back.
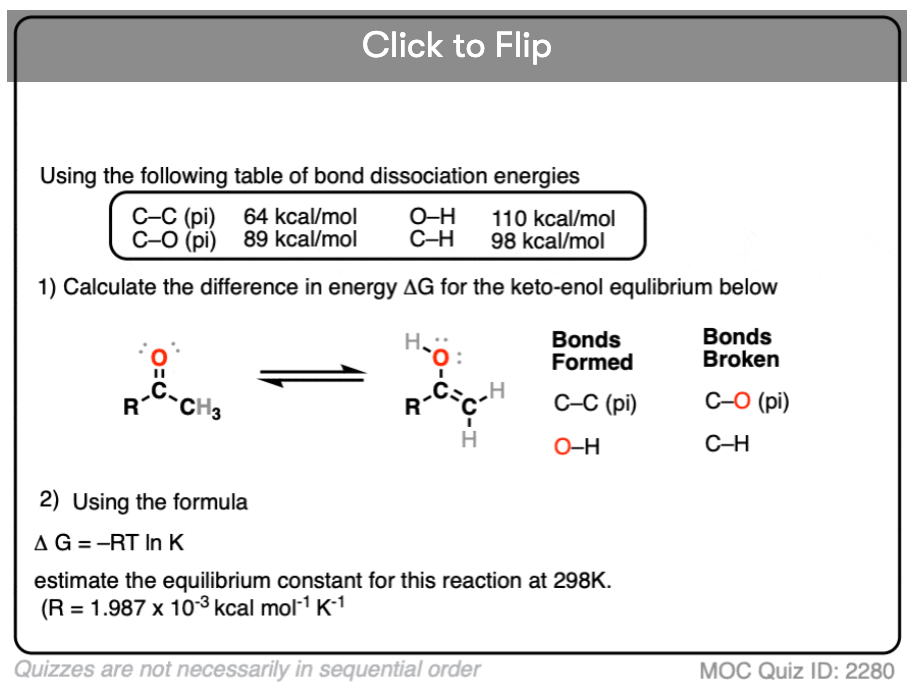
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
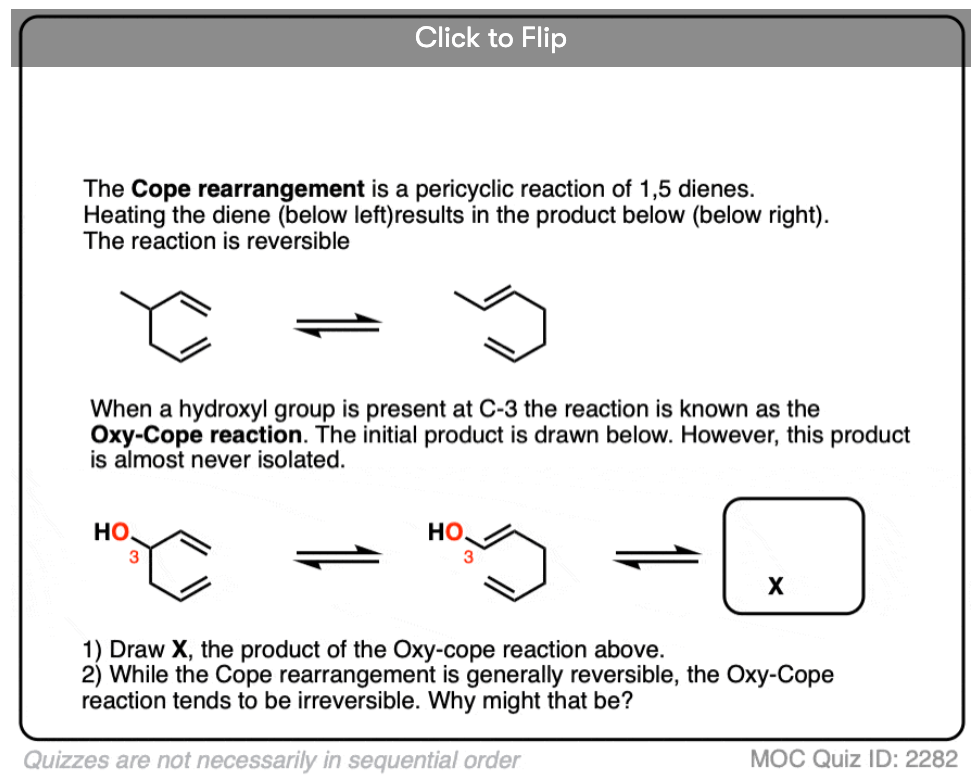
Become a MOC member to see the clickable quiz with answers on the back.
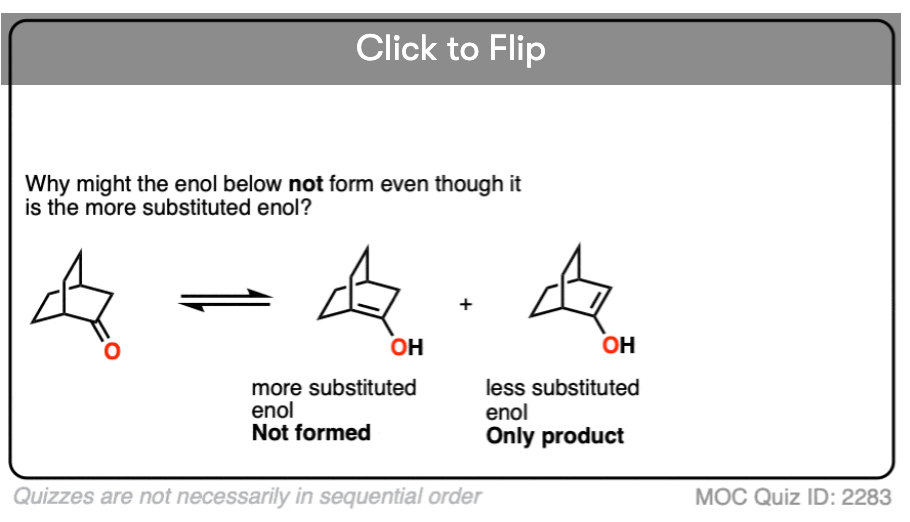
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
For a historical perspective on tautomerism, Structure and Mechanism in Organic Chemistry by C. K. Ingold is helpful.
For example, Ingold points to the coining of the term “tautomerism” by Laar (Berichte, 1885, 648 and Berichte, 1886, 19, 730) but mentions that Laar did not believe the keto and enol forms were separate species. [https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/cber.188601901165]
This was later conculsively established by separation of ethyl acetoacetonate into its ketonic and enolic components, which involved crystallizing the keto form at –80°C and showing it could be converted to the keto form (Berichte 1911, 44, 1138). Distillation of the pure (and more volatile) enol form was reported later (Berichte, 1921, 54, 579).
1. Mechanism of acid-catalyzed enolization of ketones
Gustav E. Lienhard and Tung-Chia Wang
Journal of the American Chemical Society 1969 91 (5), 1146-1153
DOI: 10.1021/ja01033a019
Study on the keto-enol equilibria of cyclohexanone supports that the rate limiting step for acid-catalyzed enol → keto tautomerism is protonation of the beta-carbon (based on similarity of rates to those for hydrolysis of enol ethers). Rate-determining step for keto → enol tautomerism is deprotonation of carbon.
2. Tautomeric equilibria in acetoacetic acid
Karen D. Grande and Stuart M. Rosenfeld
The Journal of Organic Chemistry 1980 45 (9), 1626-1628
DOI: 10.1021/jo01297a017
From the abstract: “Tautomeric equilibria in acetoacetic acid have been examined by 1H NMR and found to be strongly solvent dependent. Values for enol tautomer range from less than 2% in D2O to 49% in CCl4. Chemical shift data suggest that the enol tautomer is internally hydrogen bonded in the less polar solvents and that internal hydrogen bonding is unimportant for the keto tautomer.”
3. Generation of simple enols in aqueous solution from alkali metal enolates. Some chemistry of isobutyraldehyde enol
Y. Chiang, A. J. Kresge, and P. A. Walsh
Journal of the American Chemical Society 1986 108 (20), 6314-6320
DOI: 10.1021/ja00280a032
Gives an estimate for the equilibrium constant for keto-enol interconversion of isobutyraldehyde as K= 1.37 × 10-4 , and estimates the pKa of the enol form as 11.63 and the pKa of the keto form as 15.49.
4. Kinetics and thermodynamics of keto-enol tautomerism of simple carbonyl compounds: an approach based on a kinetic study of halogenation at low halogen concentrations
Jacques Emile Dubois, Mohiedine El-Alaoui, and Jean Toullec
Journal of the American Chemical Society 1981 103 (18), 5393-5401
DOI: 10.1021/ja00408a020
Contains rate and equilibrium constants for the enolization of various ketones in aqueous solution (see Table 2, p. 5396), particularly cyclic alkanones and substituted acetophenones.
5. Solvent effects on keto-enol equilibria: tests of quantitative models
Sander G. Mills and Peter Beak
The Journal of Organic Chemistry 1985 50 (8), 1216-1224
DOI: 10.1021/jo00208a014
Interesting study on several different keto-enol tautomer pairs. Interesting note from the abstract: “In general, for the isomer pairs in which the enol cannot form an internal hydrogen bond, the equilibria appear to be controlled almost completely by the hydrogen-bonding basicity of the solvent” (emphasis mine).
6. Microwave spectroscopic study of malonaldehyde (3-hydroxy-2-propenal). 2. Structure, dipole moment, and tunneling
Steven L. Baughcum, Richard W. Duerst, Walter F. Rowe, Zuzana Smith, and E. Bright Wilson
Journal of the American Chemical Society 1981 103 (21), 6296-6303
DOI: 10.1021/ja00411a005
Malonaldehyde is the simplest beta-carbonyl aldehyde. This study uses microwave spectroscopy to ascertain the bond lengths in the internal hydrogen-bonded structure of the malonaldehyde enol tautomer.
7. Gas-phase acidities and heats of formation of 2,4- and 2,5-cyclohexadien-1-one, the keto tautomers of phenol
Christopher S. Shiner, Paul E. Vorndam, and Steven R. Kass
Journal of the American Chemical Society 1986 108 (19), 5699-5701
DOI: 10.1021/ja00279a006
The authors made the two keto tautomers of phenol in the gas phase (through a retro Diels-Alder reaction!) and studied their acidities and heats of formation. The heat of formation of the [2H]-tautomer of phenol is ca. 6 kcal/mol higher than that of phenol, whereas the heat of formation of the [4H]-tautomer is about 10 kcal/mol higher than phenol.
8. Superacidity and Superelectrophilicity of BF3−Carbonyl Complexes
Jianhua Ren, Christopher J. Cramer, and Robert R. Squires
Journal of the American Chemical Society 1999 121 (11), 2633-2634
DOI: 10.1021/ja9840899
The authors studied the acidity of acetaldehyde (pKa 17) complexed to boron trifluoride (BF3) and found that BF3 has an astounding impact on the acidity of acetaldehyde. Most studies were done in the gas phase, but a solution phase calculation estimates the acidity of acetaldehyde coordinated to BF3 as having a pKa of -7 ; a 24 order of magnitude increase in acidity!
hey ,look at C vs D in the last table . Why does substitution decrease the enol content . It should stabilize enol more . I checked the values of 3 pentanone and acetone . found out that 3 pentanone has more. (I couldn’t find value for methyl ethyl ketone)
This is a great point. Thank you for bringing that up. I’m going to need to double check this table.
Why is water shown as a proton source in many different reactions – as I’d imagine the departure of OH would not happen (when something protonates the H of water) as OH is a bad leaving group?
Hi sir,
Excellent article – but I believe there may be a small error. In 4. Properties of Enols, the O in the phenol species is described as sp2 hybridised but I don’t see how that is possible.
When an atom with a lone pair is adjacent to a pi bond (such as an aromatic ring) there is generally a re-hybridization of the atom to put one of the lone pairs in a p orbital, as it improves orbital overlap. (Think of it as helping to improve resonance between the O and the pi bonds)
It’s less apparent in enols / phenol but for example with enamines, the nitrogen geometry is trigonal planar, not tetrahedral.
https://www.masterorganicchemistry.com/2018/01/16/a-hybridization-shortcut/
Very well explained!!!
Can you please clear my doubt…I wonder why the Carbon on the right side (the carbon thats attached to Methoxy group) in an acetoacetate doesnt undergo tautomerism and only the other carbon does…Is there like an effect ? Thank you in advance
In general the % of enol form is highest for aldehydes, then decreases for ketones and becomes essentially undetectable for esters.
My understanding is that in the absence of other factors (e.g. hydrogen bonding, resonance, aromaticity) favorability of the enol form is closely linked to the factors that govern the thermodynamic stability of alkenes. Basically there is hyperconjugative donation of adjacent substituents into the C-C pi* bond. Electron-withdrawing groups will lower the energy of the pi* orbital, whereas electron-donating groups (like OR) will raise the energy of the pi* orbital, making these stabilizing interactions less important and lowering the stability of the alkene (enol) form.
How does temperature govern the enol content in keto-enol tautomerism
Hi there, excellent website you have here!
I was just wondering; when an enol tautomerizes, does it form a double bond with the most or least substituted neighboring carbon?
Many thanks in advance
Can I please check which ratio of keto:enol is correct. In the first case, under point 2, the ratio is 24:76 keto:enol. In the second case (same diketone and same solvent, see Case 3), the ratio is 81:19 keto:enol. If the first ratio is correct, then the ratio given for benzene as solvent (Case 3) would also be wrong.
Any help on this would be much appreciated.
I have a question why does ฿ketotester have almost zero enol content and why does enol content dramatically increase when instead of the ester you attach a phenyl group? Isn’t there conjugation with the double bond in both the cases so why is the enol content. So less in the beta keto ester
Why When a Lewis basic group is nearby the enol form is stabilized by internal hydrogen bonding…..??? what if it is lewis acid instead of lewis base ?
Please tell me which is the most stable form of 1,3 cyclohexanedione and why
Just to clairfy, enolization and keto-enol tautomerism have the same mechanism right? Thank you!
The higher bond energies of the keto form is more stable than the lower bond energies of the enol form? That seems counter-intuitive. Is that true?
Higher bond energies means “stronger bonds”, therefore “more difficult to break”. Does that make sense?
To add onto this, would it then be correct to say that the keto form has higher bond energy than the enol form but a lower overall energy (G of keto), making it thermodynamically more stable?
what r the essential conditions for keto-enol formation,?
The ketone or aldehyde must have at least one C-H bond on one of its alpha carbons!
First of all thank you. Your article is really very good and cleared many of my doubts. Please help me with this question:1,3 cyclohexanedione readily undergoes base or acid catalysed deuterium exchange whereas Bicyclo [2,2,2] octane-2,6-dione doesn’t.
Is it because of restriction in hybridisation changes at bridgehead carbons and number of alpha hydrogen is less in the later?
Yes, I think the bridgehead carbon has some restrictions on it. I don’t remember exactly, but I guess you cannot have a charged bridgehead carbon for small bicyclo compounds
Yeah, it’s because of the Breddit’s Rule, which says that any Bicyclic compound with cycles less than 6 in size, cannot have charges or pi-bonds at the bridge head positions
Hello,
Can we isolate enol compound alone???
My product exist in enol form in acidic conditions. the bond energy favours to enol side. But when i make it neutral it will again convert to keto form.
I want to isolate my product when it is in enol form.
For that i want to try your ideas. 1) Aromatization. [please let tme know how it can be done ]
2) Conjugate addition.
How can i do these processes in my compound. Please let me know…. I do welcome replies from outside….
“harish@synthite.com”
if you react your keto compound with a base say hydride that you can get an enol plus hydrogen gas which will bubble out of solution…and according to le chateliers principle it will proceed forward to completion and you will get complete enol…..i beleive …i am not sure….. havent tried in the lab yet….but you should think abt it…it may gv you the desired product:-)
Deprotonation will give you the enolate, not the enol. He is interested in the neutral enol product. Once protonated, there will again be an equilibrium between the two forms.
please help me in prepaing for iit jee . Your site and efforts are very good. I liked it very much. I want to which reactions i should pay more attation.
open Carey and Sandberg part A and learn it!!!
Thanks – C&S A is definitely the best resource for this topic. Really comprehensive, with clear experimental examples.
This isn’t a typical Indian site to focus only on IIT-JEE or such exams.
This site is all about o-chem and how simply you can explain even the most toughest areas of o-chem. And if you learn the subject well you can tackle any problems and that’s what IITs look for and not a person who just passes exam…….so learn and enjoy o-chem